Email
Email Google Scholar
Google ScholarResearch
Current work centers on protein conformational ensembles, enhanced-sampling simulation, and physically grounded machine learning for biomolecular systems.
Peptide Conformational Ensemble Generation via Generative Model
So far, sophisticated protein structure prediction methods have emerged. However, they are primarily limited to folded proteins and do not offer comprehensive insights into equilibrium properties. In this research, I aim to develop a generative model capable of producing an equilibrium ensemble of proteins. This project is supported by JST ACT-X.
Constant-Force Steered Molecular Dynamics Simulation
We proposed the constant-force steered molecular dynamics simulation to estimate unbiased dissociation rate.
The input structures and topologies are provided on GitHub.
Our review paper for non-equilibrium molecular dynamics simulation: Iida, S. & Tomoshi, K. Free energy and kinetic rate calculation via non-equilibrium molecular simulation: application to biomolecules.
Cryptic Binding Site of Proteins
We found unique side-chain fluctuations of cryptic binding sites via molecular dynamics simulations.
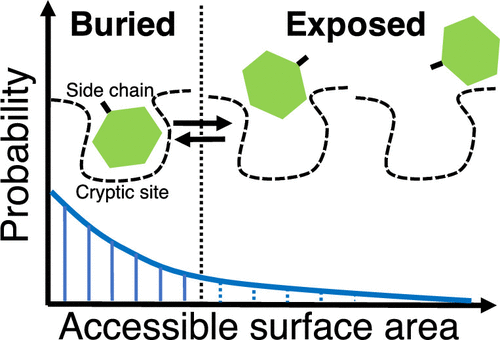
Source code to analyze the fluctuations is available on GitHub.
Intrinsically Disordered Protein
We explored the structural ensemble of an intrinsically disordered region, the p53 C-terminal domain, via a generalized-ensemble molecular dynamics simulation. We identified various binding modes on a target protein.
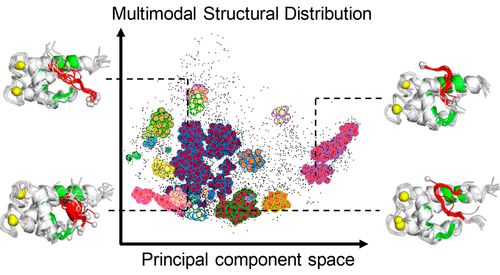
Related papers:
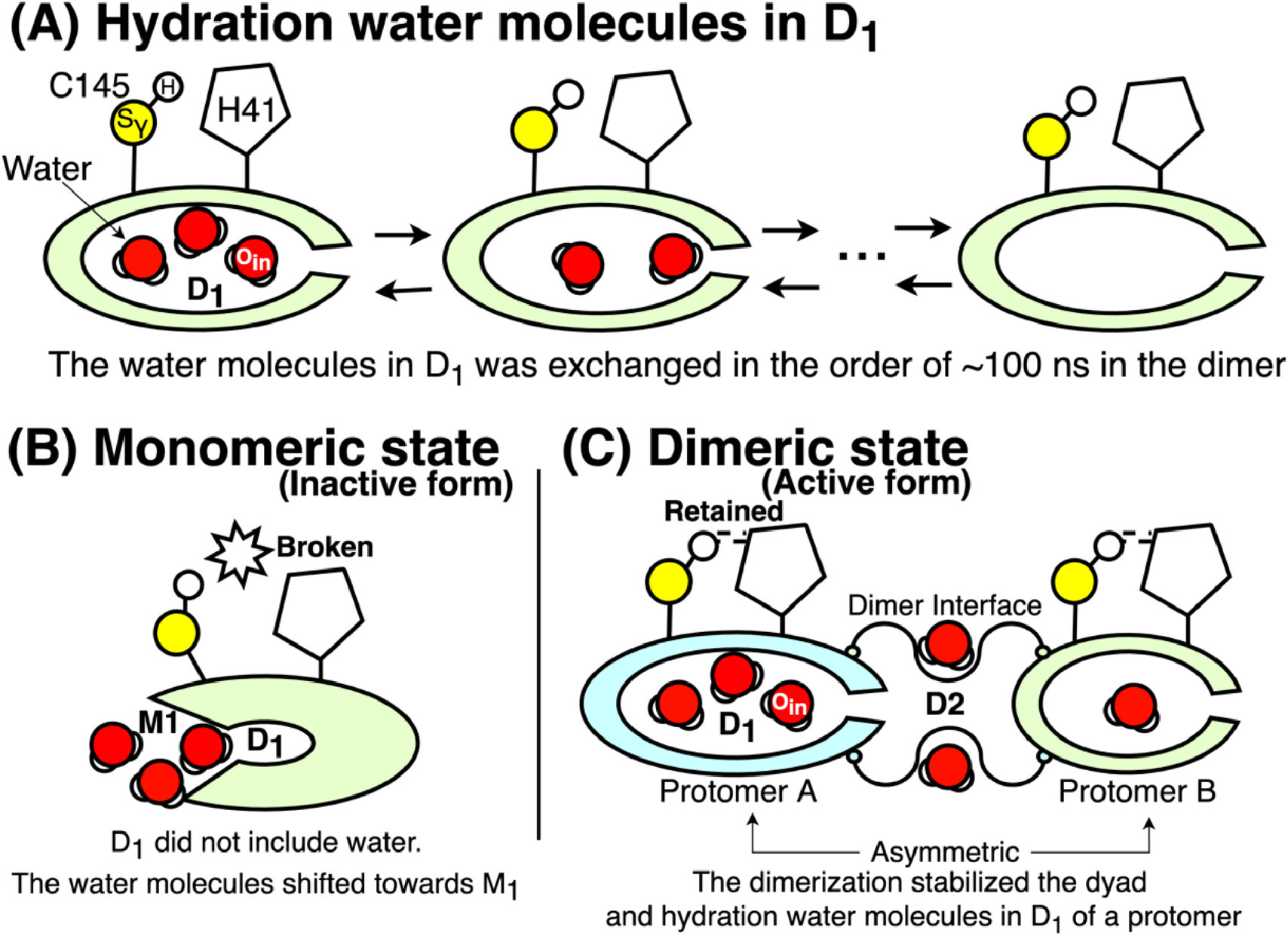
SARS-CoV-2 Main Protease
We investigated monomeric and dimeric states of the SARS-CoV-2 main protease, identifying water molecules that can be crucial for keeping the dyad configuration.